
全国咨询热线:13472751340



环己烯四醇β环氧化物(Conduritol B epoxide,简称CBE,CAS号6090-95-5)是一种重要的分子工具化合物,在溶酶体贮积症和神经退行性疾病研究中发挥着关键作用。作为一种不可逆的β-葡萄糖苷酶(β-glucosidase)抑制剂,CBE能够通过药理学方式诱导细胞和动物模型表现出戈谢病(Gaucher disease)的典型病理特征,为相关疾病的机制研究和治疗策略开发提供了有力工具。
物化性质
CBE的化学结构为1,2-脱水-肌醇,是1-L-1,2-脱水-肌醇和1-D-1,2-脱水-肌醇的外消旋混合物。其分子式为C?H??O?,分子量162.14 g/mol,IUPAC命名遵循特定的立体化学构型。
核心物化参数
● CAS号:6090-95-5
● 分子式:C?H??O?
● 分子量:162.14 g/mol
● 外观:粉末(白色至类白色)
● 溶解性:水中100 mg/mL(616.75 mM,超声助溶);DMSO中50 mg/mL(308.38 mM)
● 储存条件:2-8°C或-20°C避光保存
● 纯度标准:≥95%(TLC)或≥98%(HPLC)
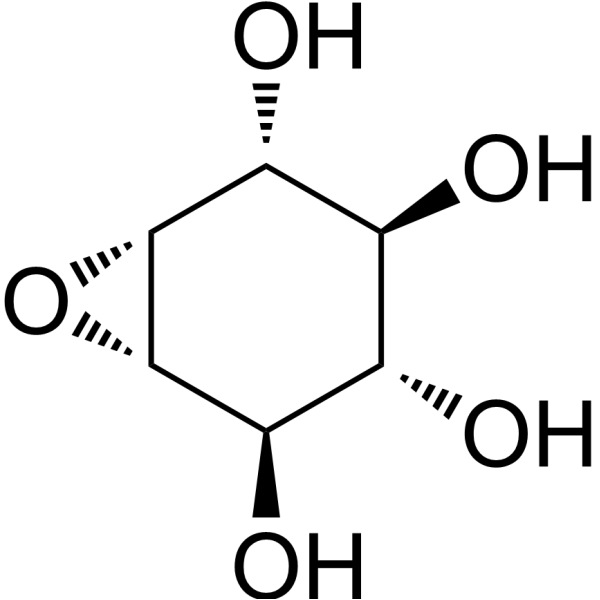 环己烯四醇β环氧化物化学结构式
环己烯四醇β环氧化物化学结构式
从外观上看,CBE呈现为白色至类白色粉末,易溶于水和二甲基亚砜(DMSO)。在实验中,常采用水或DMSO配制成储备液,需要注意的是,吸湿性DMSO可能显著影响产品的溶解度,因此建议使用新开封的DMSO溶剂。配制的储备液应分装保存,避免反复冻融造成的产品失效。
作用机制
CBE的核心作用在于其能够不可逆地抑制葡萄糖脑苷脂酶(Glucocerebrosidase,GCase)的活性。GCase是溶酶体中负责水解葡萄糖脑苷脂(glucosylceramide,GlcCer)和葡萄糖鞘氨醇(glucosylsphingosine,GlcSph)的关键酶。当CBE进入细胞后,其环氧化物结构会与GCase活性位点的催化残基发生共价结合,形成稳定的酶-抑制剂复合物,导致酶活性完全丧失[1]。
根据X射线晶体学研究,CBE在GCase活性位点通过Glu340的亲核攻击发生环氧化物开环,形成酶-环己醇酯键。值得注意的是,CBE与酶的结合不会诱导GCase的全局构象变化,但会影响活性位点入口处两个柔性环段的构象[2]。这种选择性结合机制解释了为何CBE能够作为GCase的特异性不可逆抑制剂。
当GCase活性被抑制后,细胞内会发生显著的代谢变化:葡萄糖脑苷脂和葡萄糖鞘氨醇在溶酶体内大量积累,导致溶酶体体积增大、形态异常。同时,底物积累还会引发内质网应激、线粒体功能障碍等一系列细胞病理反应,这些变化与戈谢病患者的细胞特征高度相似。
制备方法
CBE的化学合成主要采用肌醇(myo-inositol)作为起始原料,经过多步保护、酰化、脱保护和环氧化反应得到目标产物[3]。经典的合成路线可追溯至20世纪80年代中期 Michigan 大学 Radin 团队的工作。
经典合成路线概述
第一步是二醇保护反应:肌醇与环己酮在 对甲苯磺酸催化下进行缩醛保护生成1,2-单缩酮中间体。由于试剂在反应介质中溶解度较低,研究者通过原位研磨提高反应可重复性,并通过优化环己烷用量控制反应温度在110°C左右。这一步骤的产率可达85%(以回收原料校正后计)[3]。
第二步是羟基乙酰化:中间体与吡啶和乙酸酐在室温下反应生成四-O-乙酰基-1,2-O-环己叉基-肌醇。室温反应相比早期100°C条件显著降低了去缩酮化副产物的生成[3]。
第三步是缩酮水解:在酸性条件下(四比一乙酸-水,加热至100°C)将保护基团移除,生成肌醇四乙酸酯。为提高反应速率和完全度,可适当增加盐酸用量,但需注意防止过度水解[3]。
第四步是环状硫代碳酸酯形成:二醇中间体与N,N'-硫代羰基二咪唑反应生成环状硫代碳酸酯酯。
第五步和第六步分别是硫代碳酸酯移除和乙酰基团移除,得到环己烯四醇中间体。
最后一步是环氧化反应:这是合成中的关键步骤。早期方法在室温下进行环氧化需要长达6天才能完成,后经 Kishida 团队建议采用升高温度并添加自由基捕获剂的方式,将反应时间缩短至12小时[3]。
除上述路线外,以对苯醌为起始原料的三步合成法也有报道,该路线可制备(-)-环己烯四醇四乙酸酯[4]。
研究历程
CBE作为研究工具的发展历程与糖生物学和溶酶体贮积症研究的深入密切相关。回顾其研究脉络,可以看到科学界对这一化合物的认识不断深化、应用领域持续拓展的过程。
早期发现与机制研究(1980年代)
20世纪80年代中期,研究者首次系统报道了CBE对多种β-葡萄糖苷酶的不可逆抑制作用[3]。这些研究表明,CBE对哺乳动物体内负责切割葡萄糖脑苷脂的酶具有极高的亲和力。由于戈谢病正是由于该酶缺陷导致遗传代谢障碍,CBE的发现为研究这一疾病提供了重要的实验工具。
Legler等人对CBE的作用机制进行了深入研究,证实其通过与酶活性位点形成共价中间体实现不可逆抑制[3]。这些基础研究为后续CBE在疾病模型构建中的应用奠定了理论基础。
结构生物学研究(2000年代)
2003年,研究者首次报道了人源GCase与CBE复合物的X射线晶体结构,分辨率达到2.4 ?[2]。这一里程碑式的研究揭示了CBE与GCase活性位点的精确相互作用方式,证实Glu340是催化亲核试剂,并阐明了活性位点入口处两个柔性环段在底物识别中的作用。结构研究还发现,CBE结合不会诱导酶的全局构象变化,但会影响Loop 345-349和Loop 394-399的构象分布[2]。
靶点选择性研究(2010年代)
2019年发表于FEBS Journal的研究首次系统评估了CBE在体内的靶点占有率[1]。利用活性基团蛋白分析(Activity-Based Protein Profiling,ABPP)技术,研究者发现CBE在高浓度时也会抑制非溶酶体β-葡萄糖苷酶(GBA2)和溶酶体α-葡萄糖苷酶(GAA)。不过,在小鼠大脑中存在一个相对较窄但可接受的选择性窗口,可实现GBA的选择性抑制[1]。这项研究为CBE在动物模型中的合理使用提供了重要指导。
细胞模型优化(2020年代)
近年来,基于CBE处理的细胞模型研究取得新进展。在神经退行性疾病领域,CBE处理的人诱导多能干细胞(iPSC)来源的前脑神经元已成为研究GCase缺陷对轴突运输、溶酶体完整性等过程影响的重要工具[5]。研究表明,CBE处理(100 μM,持续给药)能够完全抑制GCase活性,导致GlcCer积累,但不显著影响溶酶体的轴突运输或溶酶体破裂事件[5]。
应用领域
CBE的主要应用价值在于构建戈谢病和帕金森病的药理学模型,以及在β-葡萄糖苷酶活性测定中区分不同类型的β-葡萄糖苷酶活性。
戈谢病模型构建
戈谢病是最常见的溶酶体贮积症,由GBA1基因突变导致GCase活性缺陷引起。CBE处理能够在正常细胞中模拟GCase缺陷的病理状态,包括葡萄糖脑苷脂和葡萄糖鞘氨醇的积累、溶酶体体积增大、星形胶质细胞反应性增高等特征。
动物实验中常用的给药方案包括:腹腔注射100 mg/kg/天。短期实验中,可在出生后第5天开始每日注射,持续6天;长期实验中,从出生后第15天开始每日注射,持续24或36天[6]。这些方案能够诱导出与人类戈谢病相似的病理改变,为疾病机制研究和治疗策略开发提供重要工具。
帕金森病研究
GBA1基因突变已成为帕金森病最大的遗传风险因素。研究表明,杂合子GBA1突变携带者患帕金森病的风险显著升高。CBE处理的细胞和动物模型可用于研究GCase活性降低与α-突触核蛋白聚集之间的关联机制,为探索帕金森病的发病机制提供实验依据。
血脑屏障模型构建
CBE还可用于构建体外戈谢病血脑屏障模型。研究显示,CBE处理(200 μM,72小时)能够降低人脑微血管内皮细胞(HBMEC)和星形胶质细胞的GCase活性,增加葡萄糖脑苷脂积累,从而模拟戈谢病表型[7]。这种模型可与iPSC来源的戈谢病神经元联合使用,用于评估新型治疗性蛋白跨血脑屏障的递送效率。
酶活性分析
在β-葡萄糖苷酶活性测定中,CBE可作为选择性抑制剂用于区分酸性β-葡萄糖苷酶(GBA1)和非溶酶体β-葡萄糖苷酶(GBA2)的活性。由于CBE对GBA1的抑制活性显著高于对GBA2的抑制活性,在适当浓度条件下可实现GBA1的选择性抑制[6]。
常见问题FAQ
Q:CBE处理对细胞有哪些主要影响?
CBE处理的主要效应包括:GCase活性完全抑制、葡萄糖脑苷脂和葡萄糖鞘氨醇在溶酶体内积累、溶酶体体积增大、星形胶质细胞GFAP表达上调。研究表明,CBE处理(100 μM)不影响iPSC来源神经元的轴突溶酶体运输或溶酶体完整性[5],但可能导致细胞应激反应。
Q:CBE与其他GCase抑制剂有何区别?
与结构类似的环菲醇(cyclophellitol)相比,CBE表现出更高的GBA1选择性。2019年的研究显示,环菲醇对GBA1和GBA2具有相似的亲和力,而CBE在适当浓度下可实现GBA的选择性抑制[1]。不过,两者都是不可逆的机制基础抑制剂,在模型构建中各有优势。
Q:CBE溶液如何配制和保存?
储备液配制建议:使用新开封的无水DMSO配制成50 mg/mL溶液(相当于308.38 mM),超声助溶至澄清。分装后于-80°C储存可保存6个月,-20°C储存可保存1个月。避免反复冻融。工作液建议现用现配,当天使用完毕[6]。
参考文献
[1] van der Lienden MJC, et al. In vivo inactivation of glycosidases by conduritol B epoxide and cyclophellitol as revealed by activity-based protein profiling. FEBS Journal, 2019. DOI: 10.1111/febs.14744
[2] Premkumar L, et al. X-ray structure of human acid-β-glucosidase covalently bound to conduritol-B-epoxide: implications for Gaucher disease. Journal of Biological Chemistry, 2005. 280(26): 23815-23819.
[3] Lee KJ, Boyd SA, Radin NS. Improved synthesis of conduritol B epoxide. Carbohydrate Research, 1985. 144: 148-154.
[4] Haines AH, Taylor DR. The synthesis of conduritol B tetraacetate from p-benzoquinone. Journal of the Chemical Society, Perkin Transactions 1, 1988: 979-983.
[5] Kim MJ, et al. Axonal transport of lysosomes is unaffected in glucocerebrosidase-inhibited iPSC-derived forebrain neurons. eNeuro, 2023. DOI: 10.1523/ENEURO.0079-23.2023
[6] MedChemExpress. Conduritol B epoxide product information. Product Catalog, 2024.
[7] University of Maryland Feldman Lab. Developing a Gaucher disease pharmacological model of the blood-brain barrier. Research Poster, 2021.
本文内容基于公开发表的科学研究数据,由瀚香生物收集整理,仅供科研人员参考与学术交流,不可用于个人用途。


